Running Inference Notebooks in NVIDIA BioNeMo
In this guide we are going to go over how to set up NVIDIA's BioNeMo container from NVIDIA NGC and run the Inference notebooks provided in this container. BioNeMo is an extension of the NVIDIA NeMo Megatron framework for GPU-accelerated training of large-scale, self-supervised large language models (LLMs) for the life sciences industry.
Pulling the Container
First you need to ensure that you have access to the BioNemo container. At the moment this container is not public and will be added to your NGC Private repository. You should see a page that looks like the screenshot below.
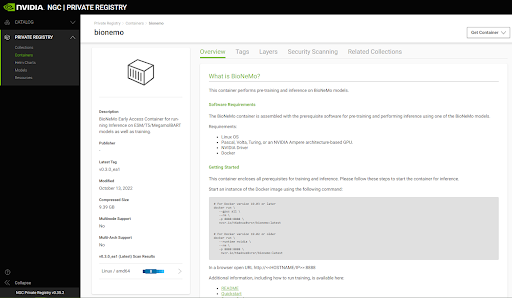
To pull this container, first access the machine you wish to install this container on and then make sure the Docker is installed. Then you will want to make sure that you have the Nvidia Container Toolkit installed. To do this, follow the instructions in the NGC documentation linked here: https://docs.nvidia.com/datacenter/cloud-native/container-toolkit/install-guide.html#installing-on-ubuntu-and-debian
Next we need to log in to the NGC container registry. The steps to do this are listed out in the Nvidia Private Registry User guide and linked here: https://docs.nvidia.com/ngc/gpu-cloud/ngc-private-registry-user-guide/index.html#accessing-ngc-registry
You should get a message saying login was successful at the end of this process. Now we are ready to pull our BioNeMo container. To do this run the command below while substituting IMAGE-TAG for the tag for the container. You can find the tag by clicking on the Get Container button on the top right of the container web page shown above.
docker pull IMAGE-TAG
The process of pulling the BioNeMo container is going to take several minutes so don’t be alarmed if it takes a long time to pull down. Once it is finished you should be able to run docker images and see the BioNeMo container listed in the output.
Running the Container
Now it’s time to run the container we pulled down. To do this run the command shown below with two substitutions. First, run docker images and copy the image ID associated with the BioNeMo image. The BioNeMo container has a few different options for models to run using the container which include esm-1nv, megamolbart, and prott5nv. You will need to choose which of these models you want to use and replace MODEL_OPTION with the name of the model.
Docker run -p 8000:8888 –gpus all IMAGE_ID MODEL_OPTION
Note that you can add -d to this command to run the container in the background. However I find it helpful to see the logs for the container when first getting it set up in case any problems arise. Once you run this command, you should be able to enter the address of the machine you are running BioNeMo on followed by :8000 in your browser and see a Jupyter Notebook instance load. An example would be 10.10.50.107:8000.
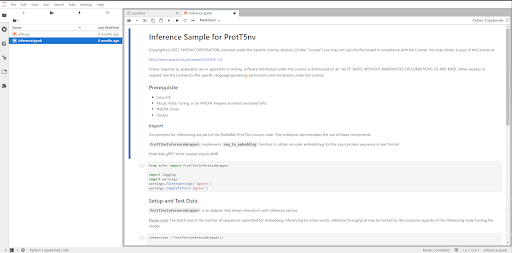
For this example I chose to launch the prott5nv model and took the screenshot below of the notebook that loaded from it.
You should be able to run inference by running the code cells in this notebook. Once you are finished, make sure to shut down your docker container using the command docker kill CONTAINER_ID once again replacing the CONTAINER_ID with the actual ID of the container image. You can find the CONTAINER_ID of your running container by running the command docker ps.