Predicting Protein Structures with Deep Learning and AlphaFold2 (on NVIDIA DGX)
In this blog post we will be looking at feeding .fasta files into AlphaFold2 running on NVIDIA DGX A100 to generate .pdb structures containing predicted protein structures.
Installation
To install AlphaFold2 follow the directions in their GitHub repository README (https://github.com/deepmind/alphafold#installation-and-running-your-first-prediction ) to pull down the AlphaFold Docker container. Note that you will need a machine running Linux with at least 3TB of storage and a large GPU (A100 on NVIDIA DGX in this case). You can get away with less space if you don’t use the full genetic database for performing your multiple sequence alignments (https://github.com/deepmind/alphafold#genetic-databases ). At the time of writing AlphaFold does not support using multiple GPUs for inference.
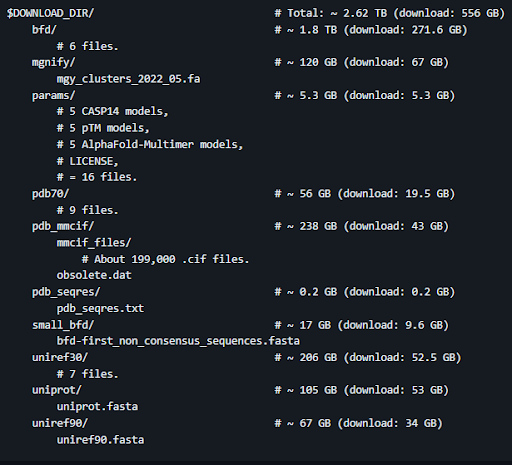
Once you have the container set up, continue following the directions to install the genetic database. For this example I installed the full database. Once your database has finished downloading, make sure it matches the structure provided in the instructions (shown below). We had some issues with the downloads not finishing on occasion so it’s a good idea to check that the file sizes match the description as well.
Examples of Running AlphaFold
To run AlphaFold we are going to run the Docker container using the command
python3 docker/run_docker.py
followed by a string of parameters. These parameters will tell the program where to find the genetic databases for the MSA and important information about what kind of run we will be doing.
For each of these examples you need to replace /db/ with the path to your own genetic database, set --fasta_paths to the path to the .fasta file you want to process, and change --output_dir to wherever you want the results to be stored.
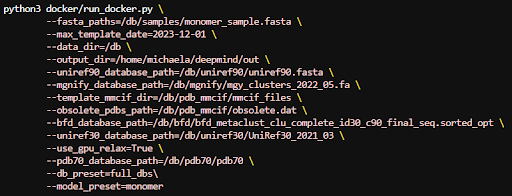
Below are a few examples of different parameter configurations for different kinds of runs. First we will be processing a monomer .fasta file and using the whole genetic database.
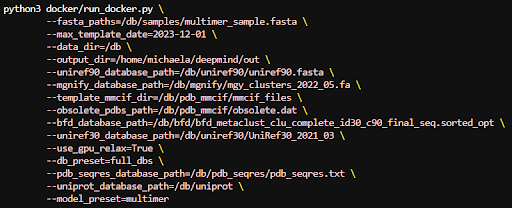
To process a multimer instead we can use parameters like the following:
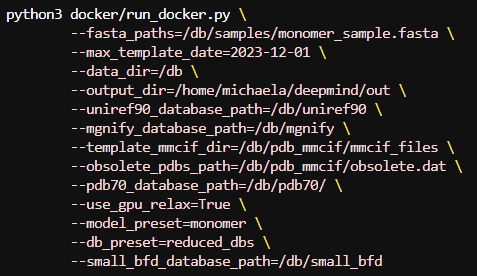
Finally to run with a reduced dataset we will need to change some of the genetic database paths. AlphaFold will throw an error if you keep any extra paths from the full database parameters so make sure to only use the ones for the reduced run shown below:
Results
Once you finish your run you should see the results in the location specified by --output_dir. One directory is created per run. Inside the directory for your run you should see something like this.
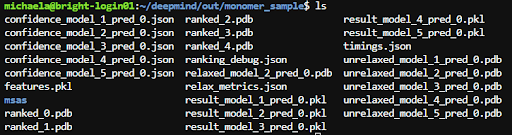
AlphaFold will give you 5 "ranked" structures with 0 being the one it is most confident about. Then you have unrelaxed versions of the ranked structures and a relaxed .pdb of the best ranked structure. Relaxation is an algorithm used at the end to make sure the structure conforms to known peptide bond geometry rules, ie there's no structures that blatantly couldn't exist in the predicted shape. The AlphaFold GitHub README (https://github.com/deepmind/alphafold#alphafold-output ) gives more info explaining the outputs.